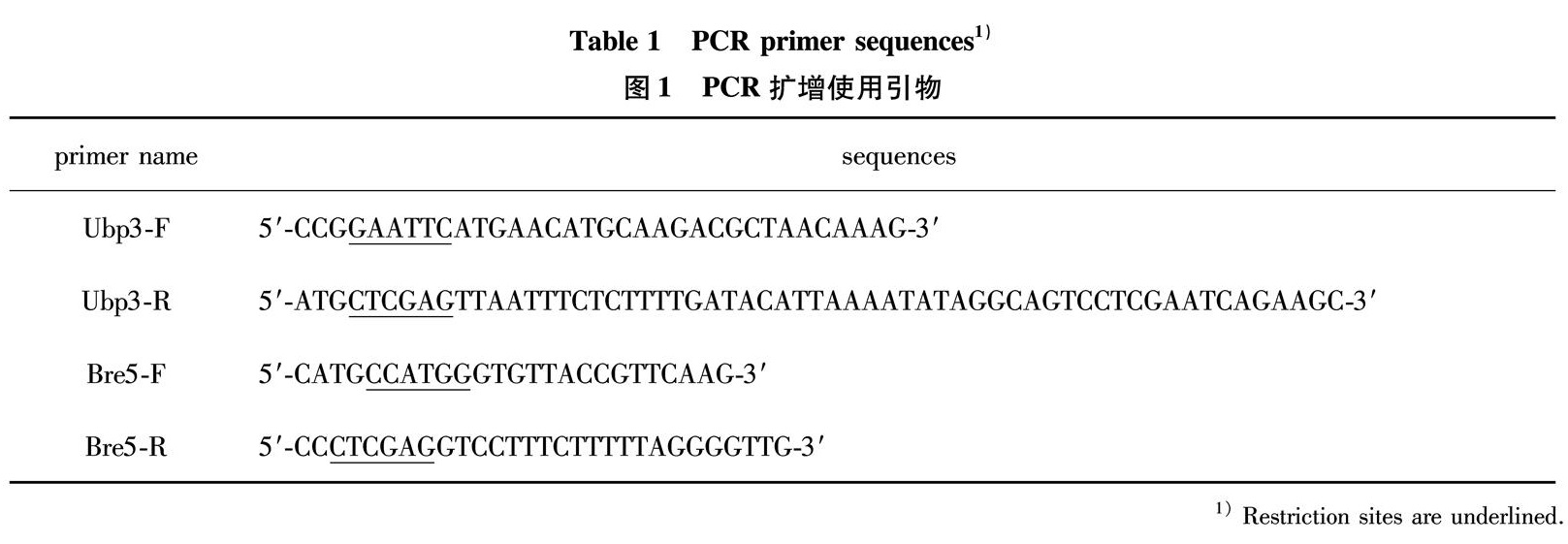
2.1 Construction of the expression plasmids
Due to the exceptional ability of GST tag to greatly enhance the solubility and stability of fused proteins, GST tag has been widely used for facilitating recombinant protein preparation; therefore we introduce a GST domain fused at the N-term of Ubp3. The Upb3 gene was amplified using Saccharomyces cerevisiae genome DNA as a template, a single band at about 2.8 kb was obtained, in accordance with the size of Upb3 coding region(Fig.1(a)); the encoding fragment was inserted into bacterial expression vector pGEX- 4T-1, and a positive plasmid designated as pGEX- 4T-1-Ubp3 was selected via colony PCR(Fig.1(b))and verified via DNA sequencing. The Bre 5 gene was amplified similarly, with a fragment of about 1.5 kb obtained(Fig.1(c)), the encoding fragment was inserted into pET-28a to introduce a 6×His tag at N-term of Bre5. The positive plasmid, designated pET-28a-Bre5, was selected by double restriction enzyme digestion(Fig.1(d))and confirmed via DNA sequencing.
(a)PCR amplification of Ubp3 coding region from Saccharomyces cerevisiae genome. Lane 1, DNA marker; Lane 2, PCR product.(b)Verification of expression plasmids pGEX- 4T-1-Ubp3 by colony PCR. Lane 1, DNA marker; Lane 2-3, PCR amplified fragments verifying two positive clones.(c)PCR amplification of Bre5 coding region from a previously constructed plasmid pET-32-Bre5. Lane 1, DNA marker; Lane 2, PCR product.(d)Verification of expression plasmids pET-28a-Bre5 by restriction enzyme digestion. Lane 1, DNA marker; Lane 2-3, two positive clones digested with Nco I and Xho I.
Fig.1 Ubp3 and Bre5 coding fragments amplified by PCR and verification of recombinant expression plasmids
图1 PCR扩增Ubp3和Bre5编码片段及质粒构建验证
2.2 Expression and purification of Ubp3
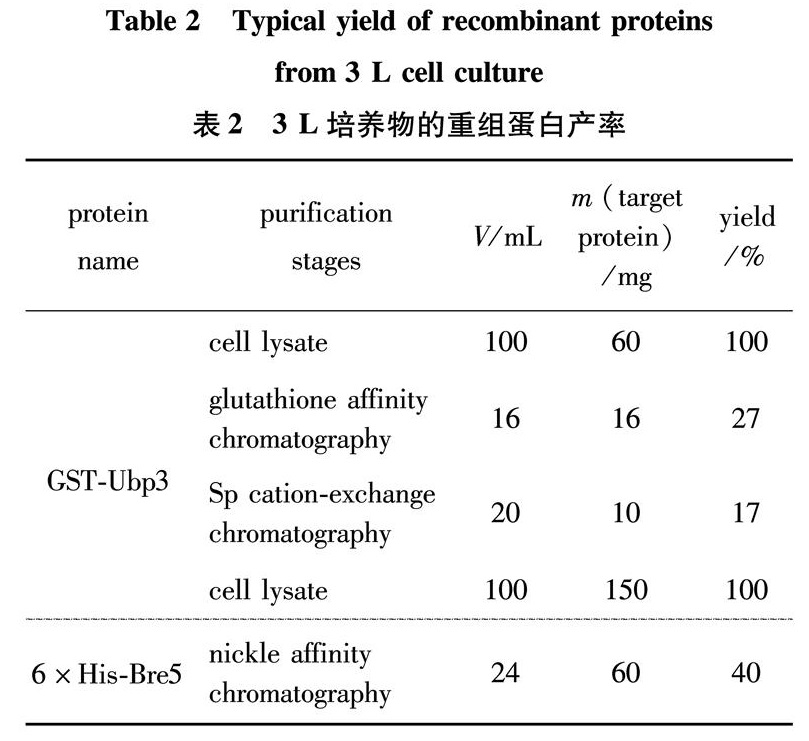
Small scale expression trials of Ubp3 in E. coli trx(DE3)were performed at various induction conditions. Compared to non-induced condition, one band migrating at about 130 kDa became apparent after Isopropyl β-D-1-thiogalactopyranoside(IPTG)induction with 0.2 mmol/L IPTG at 16 ℃, which corresponds to the GST-Ubp3 fusion, thus resulted in a relatively higher solubility of the induced protein(Fig.2(a)); therefore, we chose the same induction condition for large scale preparation. As shown in Fig.2(b), after a single-step glutathione column purification, GST-Ubp3 was enriched in elution fractions. However, the same fractions also contained several contaminating components of diverse molecular weight. The size of a prominent contaminant was about 26 kDa(Fig.2(b), Lane 6), similar to an intact GST domain, which is not surprising since GST truncates are frequently co-purified with GST fusion proteins, especially when the fused partner contains degradation-prone areas. Other major contaminating proteins appeared to be the degradation intermediates of Ubp3, since these bands remained rather unstable, almost disappeared during dialysis(compared Fig.2(b), Lane 6 with Fig.2(c), Lane 1). To further improve the purity of combined GST-Ubp3 pool and especially to remove GST truncates, we continued with ion-exchange chromatography. Based on the estimated pIs for Ubp3 and GST(7.9 for Ubp3 versus 4.5 for GST), SP sepharose was selected, and the efficacy of contaminant removal was shown in Fig.2(c). GST truncates in dialysis buffer remained poorly bound to SP resin clearly hence largely existed in flowthrough(Fig.2(c), Lane 2); in contrast, most GST-Ubp3 adsorbed to SP resin at the same condition, and was able to be efficiently eluted when NaCl concentration was increased to 0.2- 0.3 mol/L(Fig.2(c), Lane 6-9). Recovered full-length GST-Ubp3 exhibited significant improvement on purity(>85%), its identity was uniquely verified by LC-MS/MS(Fig.3). Total yields of recombinant GST-Ubp3 are listed in Table 2.
(a)Small scale expression trials of GST-Ubp3 induced at 16 ℃(Lane 1- 4)and 25 ℃(Lane 5-8)respectively. Lane 1 and 5, total proteins of uninduced cells; Lane 2 and 6, total proteins of induced cells; Lane 3 and 7, soluble fraction of induced cells; Lane 4 and 8, insoluble fraction of induced cells.(b)GST-Ubp3 purification through glutathione column chromatography. Lane 1, soluble fraction of induced cells; Lane 2, flowthrough; Lane 3- 4, wash; Lane 5-8, elution fraction.(c)Further purification of GST-Ubp3 through SP cation-exchange chromatography. Lane 1, dialyzed GST-Ubp3 pool from glutathione column chromatography; Lane 2, flowthrough; Lane 3, wash; Lane 4-6, 0.2 mol/L NaCl elution fraction; Lane 7-9, 0.3 mol/L NaCl elution fraction; Lane 10, 0.4 mol/L NaCl elution fraction.
Fig.2 SDS-PAGE analysis on expression and purification of Ubp3 in E.coli
图2 蛋白电泳分析 Ubp3在大肠杆菌内的表达及纯化
Amino acid sequence corresponding to Ubp3 was shown, with identified unique peptides highlighted in gray.
Fig.3 (Color online)Identity verification of purified Ubp3 via tandem MS/MS
图3 MS/MS鉴定纯化的Ubp3蛋白
Table 2 Typical yield of recombinant proteins from 3 L cell culture
表2 3 L培养物的重组蛋白产率
2.3 Expression and purification of Bre5
In experiments parallel to Ubp3, expression trials of Bre5 in E.coli BL21(DE3)were also performed. Upon induction, one protein with a molecular weight of about 70 kDa appears(Fig.4(a)), which is larger than the expected size of 6×His-Bre5(about 58 kDa); this is most likely due to unusual mobility of Bre5 in SDS-PAGE, since the identity of purified protein was confidently verified as Bre5 by LC-MS/MS(Fig.5). Target protein induced with 0.1 mmol/L IPTG at 16 ℃ exhibited relatively better solubility(Fig.4(a)), same induction condition was also applied to large scale purification. 6×His-Bre5 was prepared according to standard one-step nickel affinity chromatography procedure, as shown in Fig.4(b). The imidazole elution fractions were pooled and dialyzed, total yields of recombinant 6×His-Bre5 are summarized in Table 2.
(a)Small scale expression trials of 6×His-Bre5 induced at 16 ℃(Lane 1- 4)and 25 ℃(Lane 5-8)respectively. Lane 1 and 5, total proteins of uninduced cells; Lane 2 and 6, total proteins of induced cells; Lane 3 and 7, soluble fraction of induced cells; Lane 4 and 8, insoluble fraction of induced cells.(b)6×His-Bre5 purification through nickel affinity chromatography. Lane 1, soluble fraction of induced cells; Lane 2, flowthrough; Lane 3, wash; Lane 4, 50 mmol/L imidazole elution fraction; Lane 5- 6, 80 mmol/L imidazole elution fraction; Lane 7-9, 100 mmol/L imidazole elution fraction; Lane 10-12, 250 mmol/L imidazole elution fraction; Lane 13, 500 mmol/L imidazole elution fraction.
Fig.4 SDS-PAGE analysis on expression and purification of Bre5 in E.coli
图4 蛋白电泳分析Bre5蛋白在大肠杆菌内的表达及纯化
Amino acid sequence corresponding to Bre5 was shown, with identified unique peptides highlighted in gray.
Fig.5 (Color online)Identity verification of purified Bre5 via tandem MS/MS
图5 MS/MS鉴定纯化的Bre5蛋白
2.4 Functional test of recombinant Ubp3 and Bre5
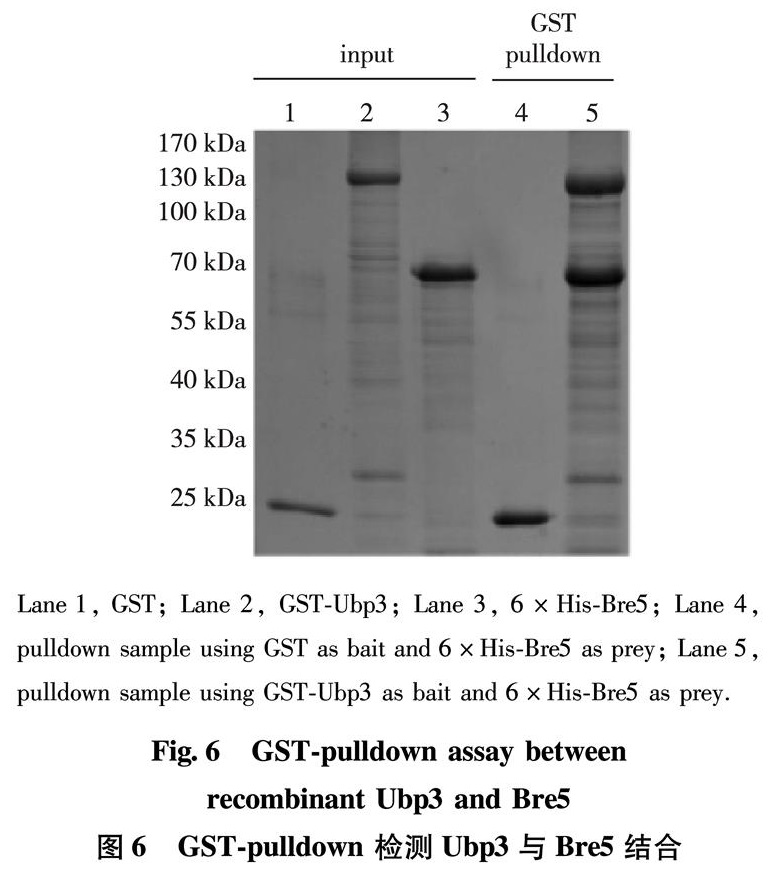
Having successfully obtained soluble Ubp3 and Bre5 in high purity, we sought to confirm whether they are properly folded or not. Previously, it has been well established that Ubp3 and Bre5 physically interact with each other in vivo and in vitro[12, 14-15]; thus, we performed a GST-pulldown to directly examine the interaction. As shown in Fig.6, in contrast to GST(Lane 4), GST-Ubp3 displays a stoichiometric interaction with Bre5(Lane 5), consistent with previous reports[14-15]. Based on these data, we conclud that our prepared recombinant Ubp3 and Bre5 are functional.
Lane 1, GST; Lane 2, GST-Ubp3; Lane 3, 6×His-Bre5; Lane 4, pulldown sample using GST as bait and 6×His-Bre5 as prey; Lane 5, pulldown sample using GST-Ubp3 as bait and 6×His-Bre5 as prey.
Fig.6 GST-pulldown assay between recombinant Ubp3 and Bre5
图6 GST-pulldown 检测Ubp3与Bre5结合
2.5 Large-scale preparation of Ubp3/Bre5 complex
The successful purification of functional Ubp3 and Bre5 individually prompted us to try preparing Ubp3/Bre5 complex directly, which is essential for further functional and structural characterization. Initially, we took our effort on co-expressing pGEX- 4T-1-Ubp3 and pET-28a-Bre5 in E.coli. Unfortunately the induction of Ubp3 and Bre5 is not at comparable level(Fig.7(a)), GST-Ubp3 induction is nearly undetectable), preventing productive complex assembly in vivo. To solve this problem, we developed a‘hybrid'procedure as shown in Fig.7(b), GST-Ubp3 was purified according to the established two-step procedure described above, except that after GST-Ubp3 binding to glutathione column, sufficient amount of recombinant 6×His-Bre5 was added to trigger the on-column complex assembly. By following this strategy, the glutathione elution fractions displayed nearly stoichiometric distributions of Ubp3 and Bre5, indicating successful complex formation on glutathione column(Fig.7(c)). Moreover, successive SP cation-exchange chromatography significantly enhanced the purity of Ubp3/Bre5 complex by efficiently removing GST truncates and other contaminants, a procedure comparable to individual Ubp3 purification(Fig.7(d)); interestingly, the preformed Ubp3/Bre5 complex well tolerated high salt elution condition(0.4- 0.5 mol/L NaCl), presenting exceptional stability.
(a)Small-scale co-expression trials of GST-Ubp3 and 6×His-Bre5 induced at 16 ℃(Lane 1- 4)and 25 ℃(Lane 5-8)respectively. Lane 1 and 5, total proteins of un-induced cells; Lane 2 and 6, total proteins of induced cells; Lane 3 and 7, soluble fraction of induced cells; Lane 4 and 8, insoluble fraction of induced cells.(b)Procedure for two-round GST-Ubp3/6×His-Bre5 complex preparation.(c)The first round of GST-Ubp3/6×His-Bre5 complex preparation through glutathione column chromatography. Lane 1, soluble fraction of induced cells; Lane 2, flowthrough after 6×His-Bre5 incubation with glutathione column; Lane 3-7, wash; Lane 8-11, elution fraction.(d)The second round of GST-Ubp3/6×His-Bre5 complex purification through SP cation-exchange chromatography. Lane 1, dialyzed GST-Ubp3/6×His-Bre5 pool from glutathione column chromatography; Lane 2, flowthrough; Lane 3- 4, wash; Lane 5, 0.2 mol/L NaCl elution fraction; Lane 6-7, 0.3 mol/L NaCl elution fraction; Lane 8-10, 0.4 mol/L NaCl elution fraction.
Fig.7 Preparation of recombinant Ubp3/Bre5 complex
图7 重组Ubp3/Bre5复合体的制备
2.6 Assessment of interactions between Ubp3 and Bre5 with Cdc48
Recently, the Ubp3/Bre5 complex has been linked to the ATPase associated with a variety of cellular activities(AAA ATPase)Cdc48. Ossareh-Nazari et al[16] proposed a direct interaction between Ubp3 and Bre5 with Cdc48 respectively, hence providing further evidence that Cdc48 has close crosstalk with deubiquitinating pathways. Taking advantages of Ubp3, Bre5 and Ubp3/Bre5 preparations, we directly examined the proposed interactions by performing a series of GST-pulldown experiments. To our surprise, we could hardly observe any interaction between GST-Ubp3 and Cdc48(Fig.8(a), Lane 5), consistently. GST-Cdc48 also failed to pulldown Bre5(Fig.8(b), Lane 3); these results are obviously contradictory to the former finding from Ossareh-Nazari et al[16]. Intriguingly, however, when the preformed GST-Ubp3/Bre5 complex was used as pulldown bait, a significant binding of Cdc48 was observed(Fig.8(c), Lane 3), indicating the assembled Ubp3/Bre5 complex is indeed able to physically interact with Cdc48. At this moment, we could not explain the discrepancy, but our results strongly suggest that more careful experiments need to be performed to uncover the real Ubp3/Bre5-Cdc48 interaction mode.
(a)GST-Ubp3 fails to interact with 6×His-Cdc48. Lane 1, 6×His-Bre5; Lane 2, 6×His-Cdc48; Lane 3, pulldown sample using GST as bait and 6×His-Cdc48 as prey; Lane 4, pulldown sample using GST-Ubp3 as bait and 6×His-Bre5 as prey; Lane 5, pulldown sample using GST-Ubp3 as bait and 6×His-Cdc48 as prey.(b)GST-Cdc48 fails to interact with 6×His-Bre5. Lane 1, 6×His-Bre5; Lane 2, pulldown sample using GST as bait and 6×His-Bre5 as prey; Lane 3, pulldown sample using GST-Cdc48 as bait and 6×His-Bre5 as prey.(c)GST-Ubp3 /6×His-Bre5 complex interacts with 6×His-Cdc48. Lane 1, 6×His-Cdc48; Lane 2, pulldown sample using GST as bait and 6×His-Cdc48 as prey; Lane 3, pulldown sample using GST-Ubp3/6×His-Bre5 complex as bait and 6×His-Cdc48 as prey.
Fig.8 GST-pulldown assays between Ubp3 and Bre5 with Cdc48
图8 GST-pulldown检测Ubp3 /Bre5 与Cdc48的结合