基金项目:国家自然科学基金资助项目(61405151)
作者简介:于长丰(1962-),男(汉族),河北省武邑县人,西安工程大学教授. E-mail:yuh55@126.com
中文责编:晨 兮; 英文责编:木 南
作者简介:于长丰(1962-),男(汉族),河北省武邑县人,西安工程大学教授. E-mail:yuh55@126.com
中文责编:晨 兮; 英文责编:木 南
Yu Changfeng and Yan Xiang'anCollege of Science, Xi'an Polytechnic University, Xi'an 710048, P.R.China
molecular physics; potential energy function; diatomic molecules and ions; Rydberg-Klein-Rees(RKR)method; force constant; spectroscopic parameter
DOI: 10.3724/SP.J.1249.2014.06561
研究得到一种既适于中性双原子分子又适于带电双原子分子离子的新的解析势能函数.用8种基本类型的双原子分子——同核中性基态双原子分子O2-X3Σ-g、同核中性激发态双原子分子K2-B1Πu、同核带电基态双原子分子离子O+2-X2Πg、同核带电激发态双原子分子离子N+2-B2Σ+u、异核中性基态双原子分子PS-X2Π1/2、异核中性激发态双原子分子BaO-A1Σ、异核带电基态双原子分子离子 37ClF--X2Σ+和异核带电激发态双原子分子离子(CS)+-A2Π,通过18个算例对势能函数进行验证,并与RKR(Rydberg-Klein-Rees)实验数据进行比较,计算结果与RKR数据吻合.
A new analytic potential energy function applied to both neutral diatomic molecules and charged diatomic molecular ions is obtained. The potential energy function is examined by 18 examples for eight different basic kinds of diatomic molecules or ions——homonuclear ground-state for neutral diatomic molecule O2-X3Σ-g, homonuclear excited-state for neutral diatomic molecule K2-B1Πu, homonuclear ground-state for charged diatomic molecular ion O+2-X2Πg, homonuclear excited-state for charged diatomic molecular ion N+2-B2Σ+u, heteronuclear ground-state for neutral diatomic molecule PS-X2Π1/2, heteronuclear excited-state for neutral diatomic molecule BaO-A1Σ, heteronuclear ground-state for charged diatomic molecular ion 37ClF--X2Σ+, heteronuclear excited-state for charged diatomic molecular ion(CS)+-A2Π, etc.. It is found that, the theoretical values for the vibrational energy level of molecules calculated with the potential energy function are in high-precision consistence with RKR(Rydberg-Klein-Rees)data or experimental data.
Analytical potential energy functions are of great significance in the study of material science, molecular spectrum,reaction dynamics of atoms and molecules, vibrational and rotational energy-level structures of molecules, interactions between laser and matter, photoionization etc.[1- 6]. So far, several kinds of representative analytical potential energy functions have been proposed, such as Morse potential[7], Rydberg potential[8], Murrell-Sorbie(M-S)potential[9] and Huxley-Murrell-Sorbie(HMS)potential[10], etc.. The potentials above are valid in describing the behaviors of some individual or classificatory molecules and ions, but none are suitable for all situations. One of the best and most extensively used analytical potential energy functions is M-S potential which is applied to most of the ground-state diatomic molecules. But M-S potential is shown to be unsatisfactory in describing the excited-state diatomic molecules. In this paper, by using a cosine function as a basic potential function and, through derivations, a high precision analytical potential function is obtained, which can describe many kinds of diatomic molecules--the neutral diatomic molecules and the charged diatomic molecular ion etc..
This potential energy function is examined with 18 examples of diatomic molecules and compared with RKR data and M-S potential. It is shown that the computational precision on the vibrational energy level of molecules calculated with this potential function is superior to that of M-S potential.
analytic potential function
Suppose that basic potential function of diatomic molecular satisfies Eq.(1)[11]
V(r)=Acos[φ+arccos(Re/r)]+B(1)
where, r is inter-nuclear distance, A and B are undetermined constants, φ is the equivalent phase difference between two interacting atoms, Re is equilibrium inter-nuclear distance. From Eq.(1)and through theoretical derivations, a universal analy-tic potential function describing diatomic molecules is obtained
V(r)=(De{∑ni=0H(i)((Re)/r)2i+a((Re)/r)2n+2+b((Re)/r)2n+4+c((Re)/r)2n+6-[ / ∑ni=0H(i)(2i)+a(2n+2)+b(2n+4)+c(2n+6)](Re)/r})/(∑ni=0H(i)(2i-1)+a(2n+1)+b(2n+3)+c(2n+5))(2)[12]
where De is the dissociation energy of diatomic molecules; H(i)=(2i)!/[4i(i!)2(2i-1)]; a, b and c are undetermined parameters which can be determined with the experimental spectroscopic parameters(ωe, ωe χe, αe, Be). For example, when n=1, 3, from Eq.(2), we have
V(r)=(2De)/(1+6a+10b+14c){1/2((Re)/r)2+a((Re)/r)4+b((Re)/r)6+c((Re)/r)8-(1+4a+6b+8c)(Re)/r},(n=1)(3)
V(r)=(16De)/(19+112a+144b+176c){1/2((Re)/r)2+1/8((Re)/r)4+1/(16)((Re)/r)6+a((Re)/r)8+b((Re)/r)10+c((Re)/r)12-((15)/8+8a+10b+12c)(Re)/r},
(n=3)(4)
Examinations show that Eq.(3)or Eq.(4)can accurately describe the interaction of diatomic molecules on a larger range of equilibrium internuclear distance in calculating the vibrational energy level of molecules but there is a definite deviation between the potential values of the long-range attractive branch of potential curve and RKR data or experimental data. So in order to further ameliorate the qualities of the long-range attractive branch, it is necessary to improve the potential energy function. In this paper, through derivations, an analytical potential function which can describe the whole range of the potential curve is obtained. The potential function is as follows
V(r)=V3(r)+sgn(r-r0)V4(r)(5)
where
V3(r)=1/2[V1(a,b,c,r)+V2(a0,b0,c0,r)]+De(6)
V4(r)=1/2[V2(a0,b0,c0,r)-V1(a,b,c,r)](7)
sgn(r-r0)=(r-r0)/(|r-r0|)(8)
We use the exactly same function for the potentials V1(a, b, c, r)and V2(a0,b0,c0,r), which can be selected from one of Eq.(3)and Eq.(4). From Eq.(5)to Eq.(8), when n=1, 3 the following potential functions can be given
{V3(r)=DeC0[1/2((Re)/r)2+C1((Re)/r)4+C2((Re)/r)6+
C3((Re)/r)8-C4((Re)/r)]+De
V4(r)=DeP0[1/2((Re)/r)2+P1((Re)/r)4+P2((Re)/r)6+
P3((Re)/r)8-P4((Re)/r)]
V(r)=V3(r)+sgn(r-r0)V4(r)
(n=1)(9)
{V3(r)=8DeC0[1/2((Re)/r)2+1/8((Re)/r)4+1/(16)((Re)/r)6+
C1((Re)/r)8+C2((Re)/r)10+C3((Re)/r)12-C4((Re)/r)]+De
V4(r)=8DeP0[1/2((Re)/r)2+1/8((Re)/r)4+1/(16)((Re)/r)6+
P1((Re)/r)8+P2((Re)/r)10+P3((Re)/r)12-P4((Re)/r)]
V(r)=V3(r)+sgn(r-r0)V4(r)
(n=3)(10)
the parameters in Eq.(9)and Eq.(10)can be calculated by the following relations
{C0=(k1+k2)/(k1k2), C1=(a0k1+ak2)/(k1+k2),
C2=(b0k1+bk2)/(k1+k2), C3=(c0k1+ck2)/(k1+k2)
P0=(k1-k2)/(k1k2), P1=(a0k1-ak2)/(k1-k2),
P2=(b0k1-bk2)/(k1-k2), P3=(c0k1-ck2)/(k1-k2)(n=1,3)(11)
{C4=1/(k1+k2)[(1+4a0+6b0+8c0)k1+
(1+4a+6b+8c)k2]
P4=1/(k1-k2)[(1+4a0+6b0+8c0)k1-
(1+4a+6b+8c)k2](n=1)(12)
{C4=1/(k1+k2)[((15)/8+8a0+10b0+12c0)k1+
((15)/8+8a+10b+12c)k2]
P4=1/(k1-k2)[((15)/8+8a0+10b0+12c0)k1-
((15)/8+8a+10b+12c)k2](n=3)(13)
{k1=1+6a+10b+14c
k2=1+6a0+10b0+14c0(n=1)(14)
{k1=19+112a+144b+176c
k2=19+112a0+144b0+176c0(n=3)(15)
the undetermined parameters a, b, c can be determined with the experimental spectroscopic parameters(ωe, ωe χe, αe, Be)of diatomic molecules. But a0, b0, c0 can be obtained by solving the following equations
{V1(a,b,c,r0)=V2(a0,b0,c0,r0)
((dV1)/(dr))r=r0=((dV2)/(dr))r=r0
V2(a0,b0,c0,r)|r=8Re=De(16)
where
r0=(Re+rm)/2·δ(17)
Here, rm is the value of inter-nuclear distance when V1(a, b, c, r)+De=De. δ is a parameter for adjusting precision.
parameters to determine a, b, c
The undetermined parameters a, b and c can be determined with the experimental spectroscopic parameters(ωe, ωe χe, αe, Be)of diatomic molecules or ions. The principle of this method is, according to the relation between undetermined parameters and force constants, to obtain a, b, c by solving linear equations. The relation between force constants and spectroscopic parameters are as follows
f2=4π2μω2ec2=(hcω2e)/(2R2eBe)(18)
f3=-(3f2)/(Re)(1+(αeωe)/(6B2e))(19)
f4=(f2)/(Re2)[15(1+(αeωe)/(6B2e))2-(8ωe χe)/(Be)](20)
where the force constants at the equilibrium inter-nuclear distance can be given as follows
fm=((dmV)/(drm))r=Re(m=2,3,4)(21)
From Eq.(3)and Eq.(4), when n=1, 3, the following linear equations can be obtained
{(1+12a+30b+56c)/(1+6a+10b+14c)=(f2R2e)/(2De)
(2+32a+100b+224c)/(1+6a+10b+14c)=-(f3R3e)/(6De)
(3+62a+240b+644c)/(1+6a+10b+14c)=(f4R4e)/(24De)(n=1)(22)
{(35+448a+720b+1 056c)/(19+112a+144b+176c)=(f2R2e)/(2De)
(98+1 792a+3 360b+5 632c)/(19+112a+144b+176c)=-(f3R3e)/(6De)(n=3)(23)
(206+5 152a+11 280b+21 648c)/(19+112a+144b+176c)=(f4R4e)/(24De)
The Eq.(22)and Eq.(23)above are all linear equations, which have unique real number solutions for the undetermined parameters a, b and c. In order to compare the potential functions Eq.(9)and Eq.(10)with M-S potential, we also provide it in the following form
V(r)=-De[1+a1(r-Re)+a2(r-Re)2+
a3(r-Re)3]exp[-a1(r-Re)](24)
The relations between undetermined parameters a1, a2, a3 and force constants are as follows
{De(a21-2a2)=f2
2De(3a1a2-3a3-a31)=f3
De(3a41-12a21a2+24a1a3)=f4(25)
Although Rydberg-Klein-Rees(RKR)inverse-method is a pure theoretical method, the values of the vibrational energy level of molecules calculated with this method are extremely and exactly consistent with the experimental data. Hence, the RKR data are usually considered as the experimental data. Under the condition of no experimental data, one of the best method is to use the RKR data to examine a certain potential energy function. The method for calculating RKR data is given as follows[13]
{Zrmax(U)=(f/g+f 2)1/2+f
rmin(U)=(f/g+f 2)1/2-f
f=((BeR2e)/(ωe χe))1/2ln[((ω2e-4ωe χeU)1/2)/(ωe-(4ωe χeU)1/2)]
g=1/((4R2eBe(ωe χe)3)1/2)[αe(4ωe χeU)1/2+ /
(2ωe χeBe-αe ωe)ln[((ω2e-4ωe χeU)1/2)/(ωe-(4ωe χeU)1/2)]]
U=ωe(υ+1/2)-ωe χe(υ+1/2)2(26)
Where rmax and rmin are the maximum and minimum classical turning points of the inter-nuclear distance for a molecule vibrating with energy U, U is the value of potential energy. From Eq.(26), f(unit: cm)and g(unit: cm-1)can be determined when the spectroscopic parameters ωe, ωe χe, αe, Be are given, and then the maximum and minimum classical turning points rmax and rmin can be calculated by using Eq.(26), and the potential curve of U(r)can be plotted out.
Substituting the potential parameters in Table 4 and Table 5 into Eq.(9)or Eq.(10), a specific analytic potential energy function of diatomic molecules can be obtained. For examining potential energy functions of Eq.(9)and Eq.(10), 18 kinds of neutral diatomic molecules and charged diatomic molecular ions have ever been investigated, and the values of the vibrational energy level of molecules calculated by the potential function are compared with RKR data, M-S potential or experimental data. The experimental data on spectroscopic parameters and the potential parameters of part diatomic molecules calculated from Eq.(16)to Eq.(25)are listed in Table 1 to Table 4. The potential parameters r0, a0, b0, c0 are calculated with Eq.(16). Table 5 is calculated with Eq.(11)to Eq.(15). The vibrational energy levels and classical turning points of heteronuclear excited-state for neutral diatomic molecules AsCl-1Δ and heternuclear ground-state for charged diatomic molecular ion(CS)+-X2Σ+ calculated by Eq.(9), Eq.(10), Eq.(24)and Eq.(26)are listed in Table 6 and Table 7.
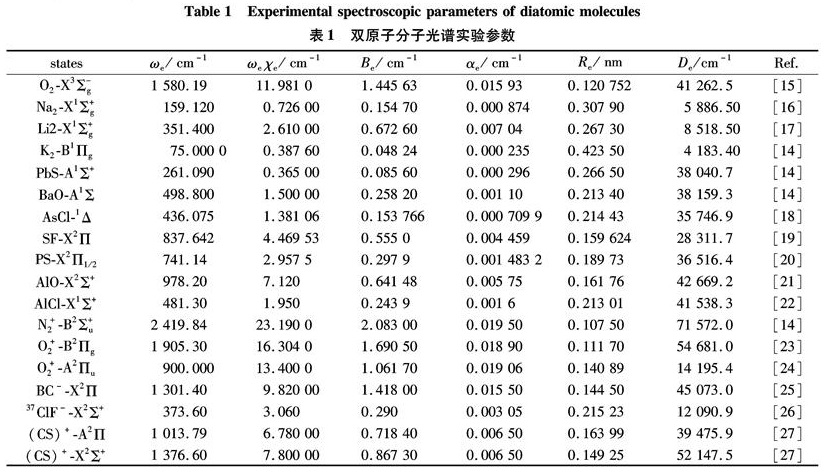
Table 1 Experimental spectroscopic parameters of diatomic molecules

Table 2 Parameters of part diatomic molecules
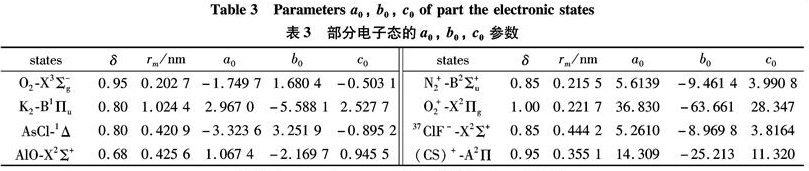
Table 3 Parameters a0, b0, c0 of part the electronic states
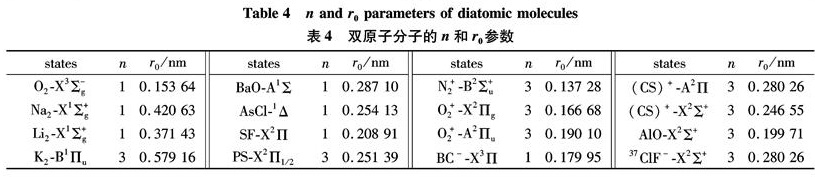
Table 4 n and r0 parameters of diatomic molecules
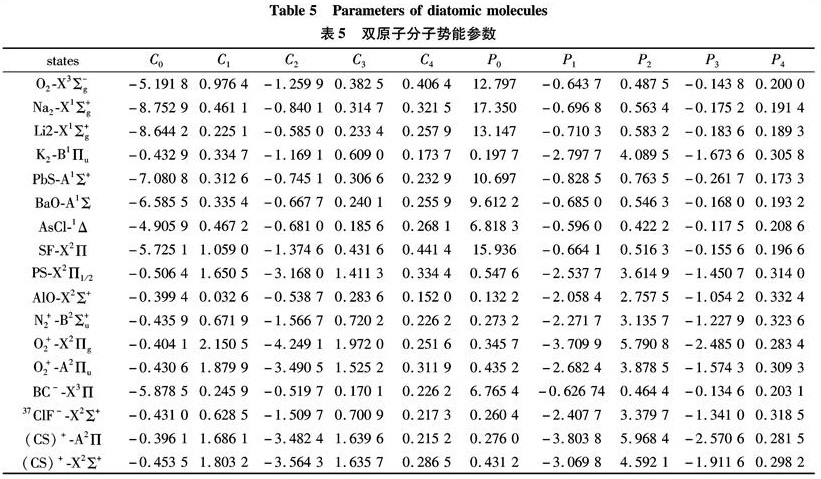
Table 5 Parameters of diatomic molecules

Table 6 The vibrational energy levels of heteronuclear excited-state for neutral diatomic molecular ion AsCl-1Δ
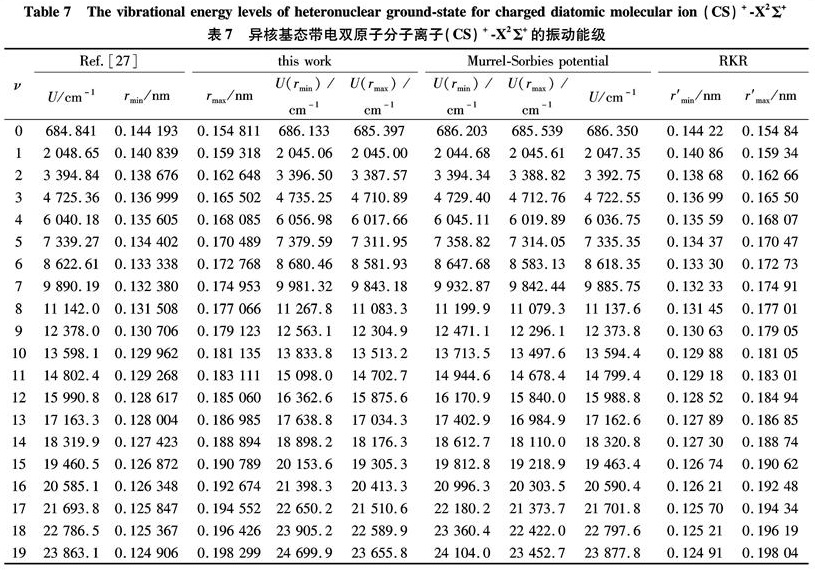
Table 7 The vibrational energy levels of heteronuclear ground-state for charged diatomic molecular ion(CS)+-X2Σ+
(Note: the relation between zero-point dissociation energy De and ground-state dissociation energy D0 given by Ref.[14] is De=D0+ ωe/2- ωe χe/4). For the limitation of the length, the computational values of the vibrational energy level of other diatomic molecules are omitted.
In order to examine the calculation precision of Eq.(9)and Eq.(10), the root-mean-square-errors(RMSE)and relative root-mean-errors between the potential values of 18 kinds of electronic states and RKR data or experimental data are listed in Table 8. For comparison, the calculation precision of M-S are also listed in Table 8. The RMSE and relative root-mean-square-errors(RRMSE)are given in the following formulas
RMSE=(1/N∑Ni=1(URKR(i)-UCAL(i))2)1/2(27)
RRMSE=(1/N∑Ni=1((URKR(i)-UCAL(i))/(URKR(i)))2)1/2(28)
The potential curves of four diatomic molecules are plotted with Origin 6.0 by using potential Eq.(9)and Eq.(10), RKR data and M-S potential are shown in Table 1.
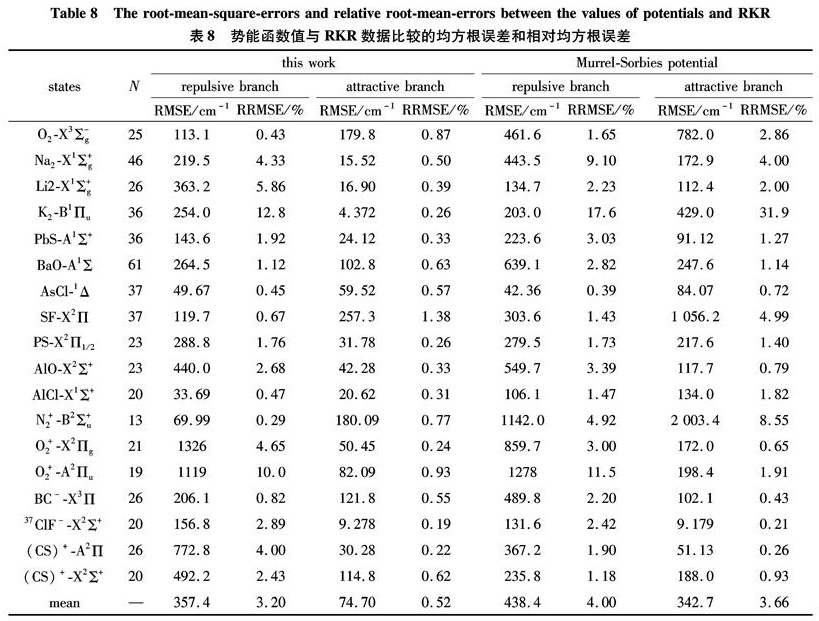
Table 8 The root-mean-square-errors and relative root-mean-errors between the values of potentials and RKR

Fig.1 Energy curves of diatomic molecules(☆=exp.; ○=RKR; —=this work; ┄=M-S)
As shown from Table 6 to Table 8, the potential function given in this paper can describe the behaviors of the whole range of the potential curve. And as for the computational precision, this potential function is superior to M-S potential.
A new analytic potential energy function which is used to describe diatomic molecules and ions is presented with a basic potential function V(r)=Acos[φ+arccos(Re/r)]+B, and good results are obtained. This potential function has two merits: ① The good universality and high computational precision. This potential is suitable for describing eight kinds of fundamental diatomic molecules——homonuclear ground-state for neutral diatomic molecules,homonuclear excited-state for neutral diatomic molecules, homonuclear ground-state for charged diatomic molecules, homonuclear excited-state for charged diatomic molecules, heteronuclear ground-state for neutral diatomic molecules, heteronuclear excited-state for neutral diatomic molecule, heternuclear ground-state for charged diatomic molecular ions, heteronuclear excited-state for charged diatomic molecular ions. ② As concerning the computational precision, this potential function is superior to M-S potential which is extensively used in atomic and molecular physics at present.
深圳大学学报理工版
JOURNAL OF SHENZHEN UNIVERSITY SCIENCE AND ENGINEERING
(1984年创刊 双月刊)
主 管 深圳大学
主 办 深圳大学
编辑出版 深圳大学学报理工版编辑部
主 编 阮双琛
国内发行 深圳市邮电局
国外发行 中国国际图书贸易集团有限公司(北京399信箱)
地 址 北京东黄城根北街16号
邮 编 100717
电 话 0755-26732266
0755-26538306
Email journal@szu.edu.cn
标准刊号 ISSN 1000-2618
CN 44-1401/N
